New DNA sequencing technologies and bioinformatics sequence analysis algorithms are able to produce high-quality chromosome-level assemblies of large genomes. However, many research communities still rely on ‘Version 1.0’ draft assemblies that are fragmented, incomplete, and lack chromosomal location data. In a study published in the journal BMC Biology, the SIB Group of Robert Waterhouse (University of Lausanne) demonstrates how genome-comparison-based evolutionary approaches can be used to help such draft assemblies along the journey towards becoming ‘finished’ reference genomes.
Exploiting conserved gene arrangements
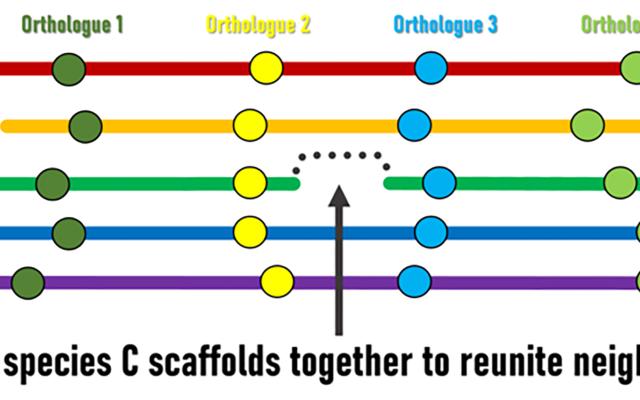
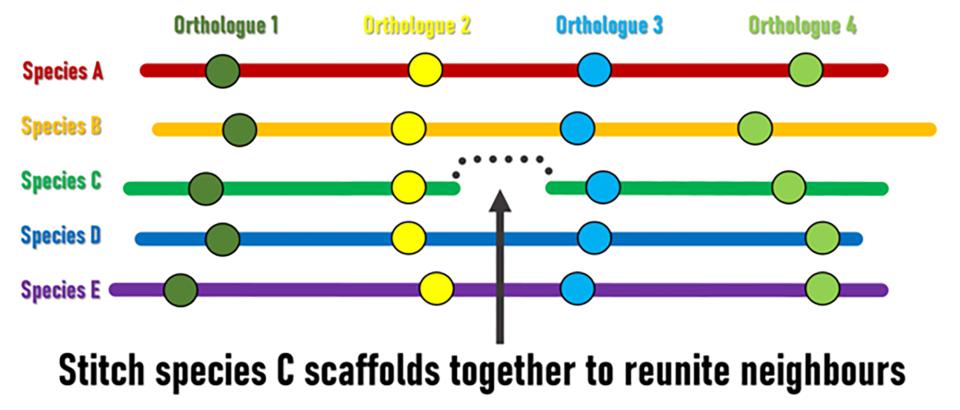
Although genomic rearrangement events lead to the shuffling of genome contents over time, regions with conserved orders and orientations in multiple species can be identified. These are known as synteny blocks, where equivalent genes in different species (orthologs) have maintained their local genomic neighbourhoods. Draft genomes are made up of genomic regions assembled into scaffolds of different lengths, but their relative orders and orientations along chromosomes is usually unknown. The SIB Researchers hypothesised that conserved synteny blocks could be used as the basis for an evolutionarily guided approach to ordering and orienting scaffolds to improve currently fragmented draft assemblies. “The logic is fairly simple,” explains lead investigator Robert Waterhouse, SIB Group Leader at the University of Lausanne’s Department of Ecology and Evolution, “when genes located at scaffold extremities in one species have orthologues from many other species that are maintained as genomic neighbours, then evolution is suggesting we can stitch those scaffolds together to reunite these pairs of genes”.
Superscaffolding and chromosome anchoring
For subsets of the assemblies the researchers integrated the synteny-based scaffold adjacencies with additional supporting data from physical mapping experiments, RNA sequencing, and extra DNA sequencing samples. The combined analyses produced 20 improved superscaffolded assemblies, where assignments of scaffolds to chromosomes spanned more than 75% of several assemblies. Chromosome anchoring of scaffolds was greatly extended for several other assemblies, and updated high-resolution cytogenetic photomaps were produced for two species. Integrating these different datasets not only allowed for enhanced superscaffolding, but also served as independent validations of the synteny-based predictions and their consensus sets.
The Waterhouse Group teamed up with researchers from George Washington University (USA) and Simon Fraser University (Canada) to apply their three independently developed bioinformatics methods, which nevertheless all employ the same basic underlying principle, to identify such ‘reunitable neighbours’ (also known as scaffold adjacencies). They tested the performance of their methods on a dataset of 21 Anopheles mosquito genomes including mostly fragmented draft assemblies. Orthologous genes, used as the genomic markers to define conserved synteny blocks, were identified using the OrthoDB orthology delineation procedure, a SwissOrthology SIB Resource.
Powering-up evolutionary inference
Leveraging the combined detection power of the three gene synteny-based methods, the analyses identified consensus sets of thousands of well-supported scaffold adjacencies that were used to build ‘superscaffolds’ (sets of stitched-together scaffolds), resulting in substantial improvements for several assemblies. While many applications in genomics research do not strictly require such high-quality assemblies, improvements in completeness, contiguity, and chromosome anchoring or assignments can substantially add to the power and breadth of biological and evolutionary inferences from comparative genomics or population genetics analyses.

Reference(s)
Waterhouse R et al. Evolutionary superscaffolding and chromosome anchoring to improve Anopheles genome assemblies. BMC Biology 2019.






